
About Me



В первые месяцы жизни дети могут выглядеть абсолютно здоровыми, хорошо прибавляют в весе. Заболевание манифестирует в возрасте 2—6 мес. Ранними симптомами являются запах плесени, исходящей от мочи и кожи ребенка (“мышиный” запах), рвота, повышенная возбудимость.
Характерны гипопигментация кожи, волос, радужки глаз. В дальнейшем нарушения миелинизации нервных волокон приводят к развитию тяжелой умственной отсталости, эпилептическим припадкам, двигательным нарушениям (пирамидному синдрому, атаксии, гиперкинезам).
Лабораторная диагностика заболевания основана на выявлении большого количества фенилаланина в крови и моче, относительно проста и возможна сразу после рождения (для ранней диагностики у новорожденных используется проба Феллинга). Если больной в течение последующей жизни соблюдает диету с резким ограничением содержания фенилаланина (в большом количестве содержится в продуктах животного происхождения, в частности в молоке), то психомоторное развитие идет нормально и никаких неврологических симптомов не развивается.
Некоторые больные, достигнув юношеского возраста, отказываются от диеты. В этом случае у них развиваются нижний спастический парапарез или умеренные психические нарушения; введение диеты может привести к обратному развитию этих симптомов.
Мой список блогов
Популярные сообщения
-
 Болезнь (лат. morbus) — это возникающие в ответ на действие патогенных факторов нарушения нормальной жизнедеятельности, работ...
Болезнь (лат. morbus) — это возникающие в ответ на действие патогенных факторов нарушения нормальной жизнедеятельности, работ... -
 ТЕРМИНАЛЬНОЕ СОСТОЯНИЕ — пограничное между жизнью и смертью состояние: обратимое угасание функций организма, предшествующее биологическ...
ТЕРМИНАЛЬНОЕ СОСТОЯНИЕ — пограничное между жизнью и смертью состояние: обратимое угасание функций организма, предшествующее биологическ... -
 ФУРУНКУЛ — острое гнойно-некротическое воспаление волосяного фолликула и окружающей его ткани, вызванное обычно золотистым, реже — белым ст...
ФУРУНКУЛ — острое гнойно-некротическое воспаление волосяного фолликула и окружающей его ткани, вызванное обычно золотистым, реже — белым ст... -
 Эритема узловатая - заболевание из группы глубоких ангиитов кожи, проявляющееся воспалительными узлами на нижних конечностях. Клиническая ...
Эритема узловатая - заболевание из группы глубоких ангиитов кожи, проявляющееся воспалительными узлами на нижних конечностях. Клиническая ... -
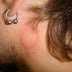 Эритразма - поверхностный псевдомикоз. Возбудитель- Corynebacterium minutissimum. Поражает верхние отделы рогового слоя эпидермиса. Источник...
Эритразма - поверхностный псевдомикоз. Возбудитель- Corynebacterium minutissimum. Поражает верхние отделы рогового слоя эпидермиса. Источник... -
 Эпидермофития - грибковое заболевание кожи. Различают паховую эпидермофитию и эпидермофитию стоп. Паховая эпидермофития. Возбудитель - Ep...
Эпидермофития - грибковое заболевание кожи. Различают паховую эпидермофитию и эпидермофитию стоп. Паховая эпидермофития. Возбудитель - Ep... -
 ФЛИКТЕНА — поражение кожи при стрептококковой пиодермии: пузырь с вялой тонкой покрышкой и серозным содержимым, которое быстро становится с...
ФЛИКТЕНА — поражение кожи при стрептококковой пиодермии: пузырь с вялой тонкой покрышкой и серозным содержимым, которое быстро становится с... -
 ТРОХАНТЕРИТ — воспаление большого вертела бедренной кости, чаще туберкулезной природы. Проявления : боль, усиливающаяся при отведении бедр...
ТРОХАНТЕРИТ — воспаление большого вертела бедренной кости, чаще туберкулезной природы. Проявления : боль, усиливающаяся при отведении бедр... -
 ТРОЙНИЧНОГО НЕРВА НЕВРАЛГИЯ (болевой тик) — клинический синдром поражения тройничного нерва: повторные короткие приступы интенсивной боли с...
ТРОЙНИЧНОГО НЕРВА НЕВРАЛГИЯ (болевой тик) — клинический синдром поражения тройничного нерва: повторные короткие приступы интенсивной боли с... -
 Энцефалит - воспаление головного мозга. Различают первичный и вторичный (на фоне какого-либо заболевания) энцефалит. К первичным относят эпи...
Энцефалит - воспаление головного мозга. Различают первичный и вторичный (на фоне какого-либо заболевания) энцефалит. К первичным относят эпи...